新型コロナウイルス [ヨコハマ]
新型コロナウイルス
心配です。
いろいろな人が集まるイベントは、中止になっているのに、通勤電車は、相変わらず満員だし。
どこに行っても、マスク売っていないし...

今回の新型ウイルスの流行では、中国は、ネット上に積極的に情報を公開しているようです。
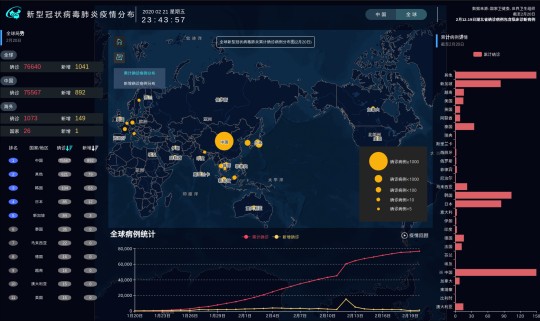
中国疾病预防控制中心 (中国疾病予防管理センター)
新型冠状病毒肺炎疫情分布 http://2019ncov.chinacdc.cn/2019-nCoV/global.html
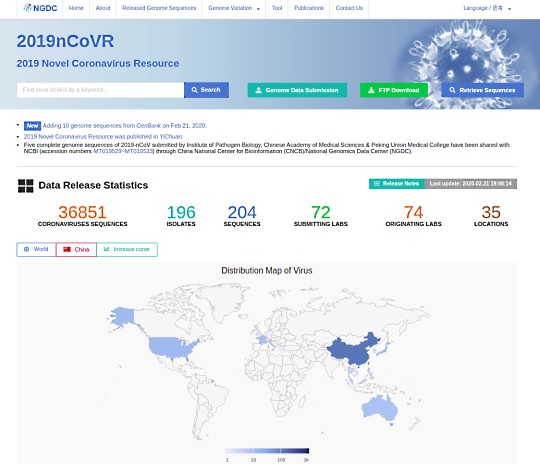
National Genomics Data Center には、各国の感染者のウイルスの遺伝子配列のデータも公開されています。
2019 Novel Coronavirus Resource
https://bigd.big.ac.cn/ncov/?lang=en
R言語のggmsa (Plot Multiple Sequence Alignment using 'ggplot2' : https://cran.r-project.org/web/packages/ggmsa/index.html )というライブラリを使って、2019 Novel Coronavirus Resourceにあるデータから新型コロナウイルスの系統樹を作ってみました。
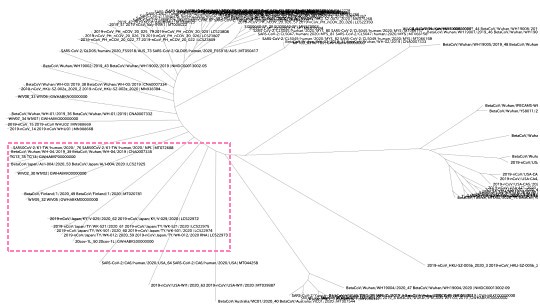
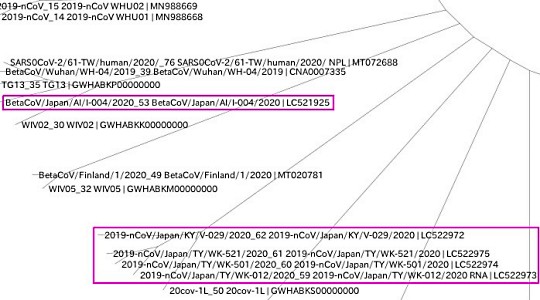
新型コロナウイルスには、遺伝子の違うグループがいくつかあって、だいたい地域ごとにグループがわかれていることがわかります。
(左下に日本で発見されたウイルスのグループがあります。)
日本のデータは、ひとつだけグループからはずれてWuhan (武漢市)で見つかったパターンに近いものがありますが、あとは、ひとかたまりになっているので、サンプルを採取した時点(いずれも1月31日以前)には、すでに国内で2次感染が広まっていたことを表しているようです。
2019 Novel Coronavirus Resource ( https://bigd.big.ac.cn/ncov/?lang=en )のデータは、シーケンスの長さが違うものや、IDが重複しているものがあったりで、そのままではggmsaで処理できなかったので、biopythonを使って、データを整形しました。
https://pypi.org/project/biopython/
心配です。
いろいろな人が集まるイベントは、中止になっているのに、通勤電車は、相変わらず満員だし。
どこに行っても、マスク売っていないし...
今回の新型ウイルスの流行では、中国は、ネット上に積極的に情報を公開しているようです。
中国疾病预防控制中心 (中国疾病予防管理センター)
新型冠状病毒肺炎疫情分布 http://2019ncov.chinacdc.cn/2019-nCoV/global.html
National Genomics Data Center には、各国の感染者のウイルスの遺伝子配列のデータも公開されています。
2019 Novel Coronavirus Resource
https://bigd.big.ac.cn/ncov/?lang=en
R言語のggmsa (Plot Multiple Sequence Alignment using 'ggplot2' : https://cran.r-project.org/web/packages/ggmsa/index.html )というライブラリを使って、2019 Novel Coronavirus Resourceにあるデータから新型コロナウイルスの系統樹を作ってみました。
library(Biostrings)
library(ape)
library(ggtree)
library(ggmsa)
sequences = "2019nCoV_20200219.fasta"
x = readAAStringSet(sequences)
d =as.dist(stringDist(x, method = "hamming")/width(x)[1])
tree = bionj(d)
p =ggtree(tree, lwd=0.1, layout="daylight",branch.length='none' )
+ geom_tiplab(align = FALSE, geom = "text", size=3)
p
新型コロナウイルスには、遺伝子の違うグループがいくつかあって、だいたい地域ごとにグループがわかれていることがわかります。
(左下に日本で発見されたウイルスのグループがあります。)
日本のデータは、ひとつだけグループからはずれてWuhan (武漢市)で見つかったパターンに近いものがありますが、あとは、ひとかたまりになっているので、サンプルを採取した時点(いずれも1月31日以前)には、すでに国内で2次感染が広まっていたことを表しているようです。
2019 Novel Coronavirus Resource ( https://bigd.big.ac.cn/ncov/?lang=en )のデータは、シーケンスの長さが違うものや、IDが重複しているものがあったりで、そのままではggmsaで処理できなかったので、biopythonを使って、データを整形しました。
https://pypi.org/project/biopython/
from Bio import Phylo
from Bio.Phylo.TreeConstruction import DistanceCalculator
from Bio import AlignIO
from Bio import SeqIO
from Bio import Seq
input_file = 'all.fasta'
records = SeqIO.parse(input_file, 'fasta')
records = list(records)
print(records[0].id)
> Wuhan-Hu-1
maxlen = max(len(record.seq) for record in records)
print(maxlen)
>29903
i = 1
for record in records:
record.id = record.id + '_' + str(i)
i += 1
if len(record.seq) != maxlen:
sequence = str(record.seq).ljust(maxlen, '.')
record.seq = Seq.Seq(sequence)
assert all(len(record.seq) == maxlen for record in records)
print(records[0])
>ID: Wuhan-Hu-1_1
>Name: Wuhan-Hu-1
>Description: Wuhan-Hu-1 | MN908947
>Number of features: 0
>Seq('ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT...AAA', SingleLetterAlphabet())
output_file = '2019nCoV_20200219.fasta'
with open(output_file, 'w') as f:
SeqIO.write(records, f, 'fasta')
alignment = AlignIO.read(output_file, "fasta")
print(alignment)
>SingleLetterAlphabet() alignment with 82 rows and 29903 columns
>ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTC...AAA Wuhan-Hu-1_1
>CAACCAACTTTCGATCTCTTGTAGATCTGTTCTCTAAACGAACT...... 2019-nCoV_HKU-SZ-002a_2020_2
>ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTC...... 2019-nCoV_HKU-SZ-005b_2020_3
>ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTC...... 2019-nCoV/USA-WA1/2020_4
>...
>TGAGTTATGAGGATCAAGATGCACTTTTCGCATATACAAAACGT...... 2019-nCoV_HKU-SZ-001_2020_17
>AATGTCTATGCAGATTCATTTGTAATTAGAGGTGATGAAGTCAG...... 2019-nCoV_HKU-SZ-002b_2020_18
>...




お久しぶりのコメントです。
・・・
35日間の入院中、ご訪問できなかったのに、nice! をいただいて、ありがとうございます。
今日退院でき、今後は通院治療ですので、ようやくPCが使えて、nice! のお返しができます!
今後ともよろしくお願いします。
by とし@黒猫 (2020-02-26 20:19)
とし@黒猫 さん
退院おめでとうございます。
病院の外は、いつのまに新型コロナウイルスとか流行ってしまっています。
どうかご自愛ください。
by aoken (2020-02-26 20:35)